Protein Genomics And Annotations
Genome projects employ state-of-the-art DNA sequencing, mapping, and computational technologies (including cross-disciplinary experimental designs) to expand our knowledge and understanding of molecular/cellular mechanisms, gene repertoires, genome architecture, and evolution. The revolution in new sequencing technologies and computational developments has allowed researchers to drive advances in genome assembly and annotation to make the process better, faster, and cheaper with key model organisms.
Such technical advantages and established recommendations and strategies have been widely applied in humans , terrestrial animals, and plants and crops . Genomic applications in aquatic species that could be potentially important for aquaculture are slower compared with human, livestock, and crops , compounded by larger diversity, lack of reference genomes, and more novice aquaculture industries. Given that aquaculture is the most rapidly expanding food sector, with the widest diversity of species cultured, it is poised for rapid adoption of genomics applications as these become more accessible. For any specific advice on application of genomics to aquaculture, please refer to previous works.
01

DNA Sequencing
DNA sequencing is the process of determining the precise order of nucleotides (the building blocks of DNA) in a DNA molecule. This technique is crucial for various applications, including genetic research, medical diagnostics, and evolutionary biology.
02

DNA Mapping
DNA mapping is the process of creating a visual representation of the locations of genes and other important features on a DNA molecule. It helps researchers understand the structure, function, and organization of genomes.
03
DNA Computational Technology
DNA computational technology involves using computational methods to analyze and interpret genomic data. Key aspects include bioinformatics for processing biological data, genome assembly to reconstruct sequences, and variant calling to identify genetic variations
Many seminal biological discoveries in the 20th century were made using only a genetic analysis of a few selected model organisms because they were readily available for genetic analysis. However, a high-quality and well-annotated genome assembly is increasingly becoming an essential tool for applied and basic research across many biological disciplines in the 21st century that can turn any organism into a model organism. Thus, securing more complete and accurate reference genomes and annotations before analyzing post-genome studies such as genome-wide association studies, structural variations, and posttranslational studies (methylation or histone modification) has become a cornerstone of modern genomics. Chromosome-level high-quality genomes (including structural and functional annotations) are differentiated from draft genomes by their completeness (low number of gaps and ambiguous Ns), low number of assembly errors, and a high percentage of sequences assembled into chromosomes.
For diploids, using a minimum 60× depth for PacBio, ONT, 60× for Illumina (San Diego, California, United States of America), and 100× for Hi-C data (Phase Genomics, Seattle, Washington, USA) (an extension of chromosome conformation capture, 3C) is recommended. High-quality end-to-end genome assembly and annotation of small eukaryotic (approximately 1 Gb diploid) and prokaryotic organisms have been achievable with small-to-medium financial resources and limited time, labor, and skill commitments. Nearly all eukaryotic genomes still represent a significant challenge for most aquatic species that have large and complex genomes and no reference genomes.
Determining amino acid composition
It is often desirable to know the unordered amino acid composition of a protein prior to attempting to find the ordered sequence, as this knowledge can be used to facilitate the discovery of errors in the sequencing process or to distinguish between ambiguous results. Knowledge of the frequency of certain amino acids may also be used to choose which protease to use for digestion of the protein. The misincorporation of low levels of non-standard amino acids (e.g. nor leucine) into proteins may also be determined. A generalized method often referred to as amino acid analysisfor determining amino acid frequency is as follows:
1.Hydrolyse a known quantity of protein into its constituent amino acids.
2. Separate and quantify the amino acids in some way.
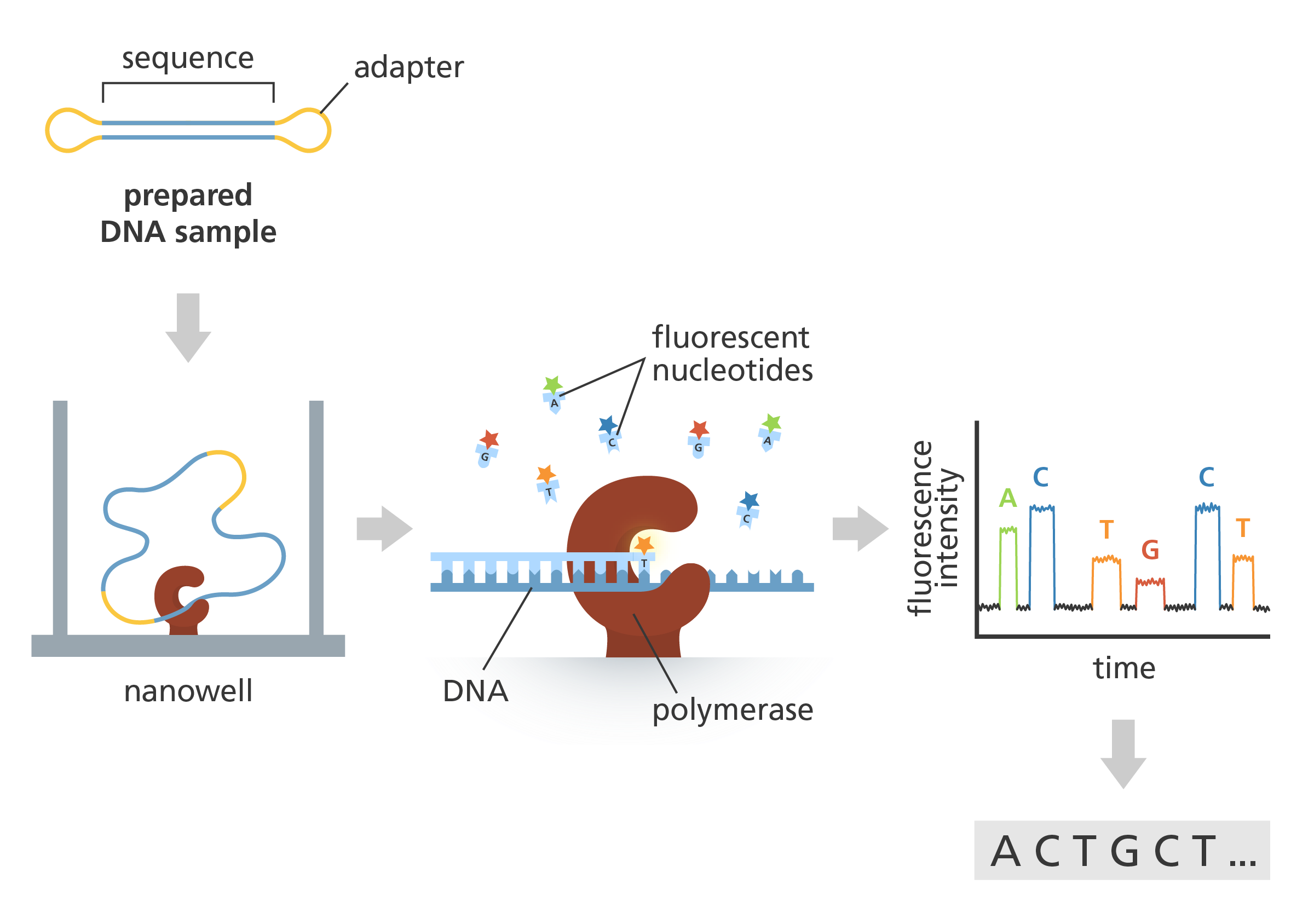
Hydrolysis
Hydrolysis is done by heating a sample of the protein in 6 M hydrochloric acid to 100–110 °C for 24 hours or longer. Proteins with many bulky hydrophobic groups may require longer heating periods. However, these conditions are so vigorous that some amino acids (serine, threonine, tyrosine, tryptophan, glutamine, and cysteine) are degraded. To circumvent this problem, Biochemistry Online suggests heating separate samples for different times, analysing each resulting solution, and extrapolating back to zero hydrolysis time. Ras tall suggests a variety of reagents to prevent or reduce degradation, such as thiol reagents or phenol to protect tryptophan and tyrosine from attack by chlorine, and pre-oxidising cysteine. He also suggests measuring the quantity of ammonia evolved to determine the extent of amide hydrolysis.
Protein sequence interpretation: a scheme new protein to be engineered in a yeast
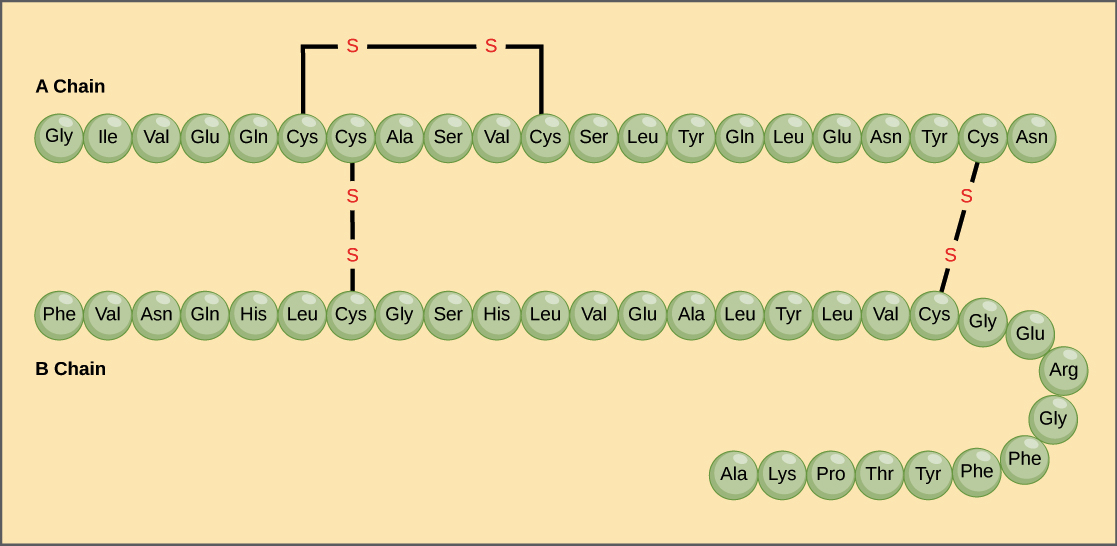
Primary structure
The simplest level of protein structure, primary structure, is simply the sequence of amino acids in a polypeptide chain. For example, the hormone insulin has two polypeptide chains, A and B, shown in diagram below. (The insulin molecule shown here is cow insulin, although its structure is similar to that of human insulin.) Each chain has its own set of amino acids, assembled in a particular order. For instance, the sequence of the A chain starts with glycine at the N-terminus and ends with asparagine at the C-terminus, and is different from the sequence of the B chain. What's up with those S-S bonds?
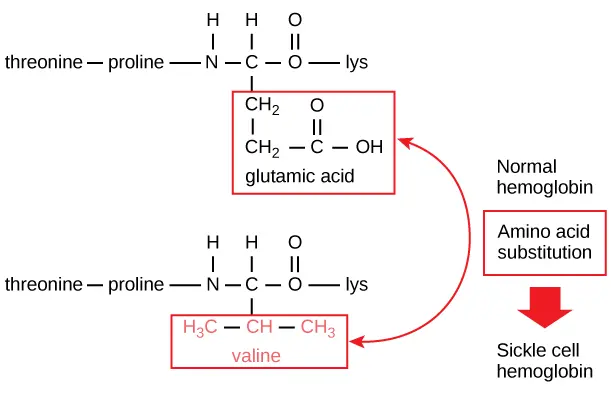
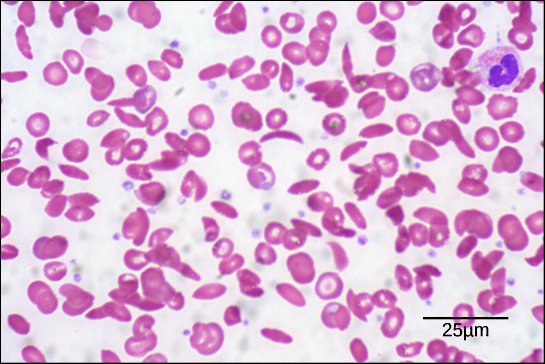
The sequence of a protein is determined by the DNA of the gene that encodes the protein (or that encodes a portion of the protein, for multi-subunit proteins). A change in the gene's DNA sequence may lead to a change in the amino acid sequence of the protein. Ever changing just one amino acid in a protein’s sequence can affect the protein’s overall structure and function. For instance, a single amino acid change is associated with sickle cell anemia, an inherited disease that affects red blood cells. In sickle cell anemia, one of the polypeptide chains that make up hemoglobin, the protein that carries oxygen in the blood, has a slight sequence change. The glutamic acid that is normally the sixth amino acid of the hemoglobin β chain (one of two types of protein chains that make up hemoglobin) is replaced by a valine. This substitution is shown for a fragment of the β chain in the diagram below.
What is most remarkable to consider is that a hemoglobin molecule is made up of two α chains and two β chains, each consisting of about 150 amino acids, for a total of about 600 amino acids in the whole protein. The difference between a normal hemoglobin molecule and a sickle cell molecule is just 2 amino acids out of the approximately 600.A person whose body makes only sickle cell hemoglobin will suffer symptoms of sickle cell anemia. These occur because the glutamic acid-to-valine amino acid change makes the hemoglobin molecules assemble into long fibers. The fibers distort disc-shaped red blood cells into crescent shapes. Examples of “sickled” cells can be seen mixed with normal, disc-like cells in the blood sample below.
Secondary structure
The next level of protein structure, secondary structure, refers to local folded structures that form within a polypeptide due to interactions between atoms of the backbone. (The backbone just refers to the polypeptide chain apart from the R groups – so all we mean here is that secondary structure does not involve R group atoms.) The most common types of secondary structures are the α helix and the β pleated sheet. Both structures are held in shape by hydrogen bonds, which form between the carbonyl O of one amino acid and the amino H of another.
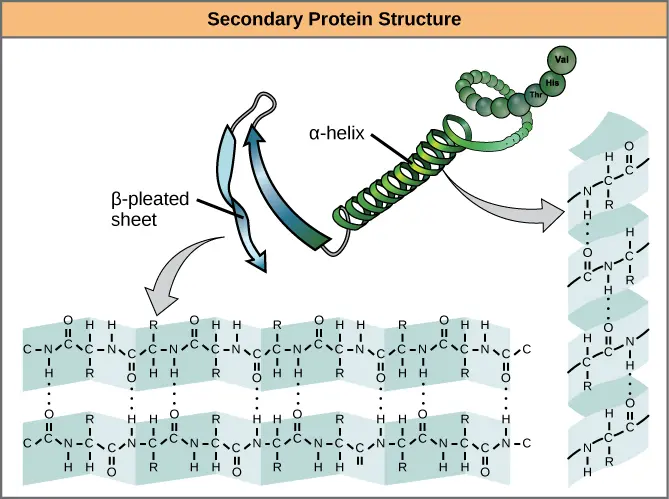
In an α helix, the carbonyl (C=O) of one amino acid is hydrogen bonded to the amino H (N-H) of an amino acid that is four down the chain. (E.g., the carbonyl of amino acid 1 would form a hydrogen bond to the N-H of amino acid 5.) This pattern of bonding pulls the polypeptide chain into a helical structure that resembles a curled ribbon, with each turn of the helix containing 3.6 amino acids. The R groups of the amino acids stick outward from the α helix, where they are free to interact\[^3\].In a β pleated sheet, two or more segments of a polypeptide chain line up next to each other, forming a sheet-like structure held together by hydrogen bonds. The hydrogen bonds form between carbonyl and amino groups of backbone, while the R groups extend above and below the plane of the sheet\[^3\]. The strands of a β pleated sheet may be parallel, pointing in the same direction (meaning that their N- and C-termini match up), or antiparallel, pointing in opposite directions (meaning that the N-terminus of one strand is positioned next to the C-terminus of the other).
Tertiary Structure
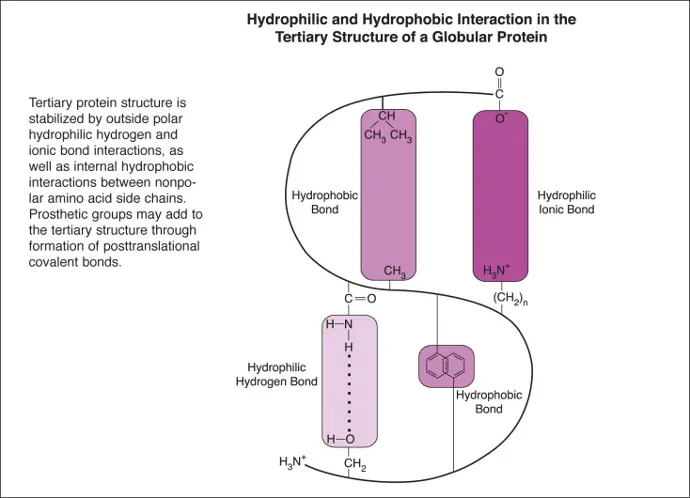
The tertiary structure of a protein refers to the overall three-dimensional arrangement of its polypeptide chain in space. It is generally stabilized by outside polar hydrophilic hydrogen and ionic bond interactions, and internal hydrophobic interactions between nonpolar amino acid side chains . Additional posttranslational covalent bonds in the tertiary structure may be formed with prosthetic groups. Tertiary folding begins while the protein is being molded into its primary polypeptide sequence. It is known to be guided through this process by chaperones, which are a family of proteins that bind hydrophobic patches and prevent them from being aggregated prematurely into nonfunctional entities.
Quaternary Structure
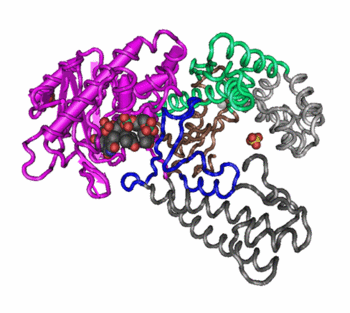
Quaternary structure in proteins is the most intricate degree of organization still considered a single molecule. To be considered to have quaternary structure, a protein must have two or more peptide chains forming subunits. The subunits can be different or identical, and in most cases they are arranged symmetrically. In general, a protein with two subunits is called a dimer; one with three subunits a trimer; and one with four subunits a tetramer.
Changes in quaternary structure can occur through conformational changes within individual subunits or through reorientation of the subunits relative to each other. It is through such changes, which underlie cooperativity and allostery in "multimeric" enzymes, that many proteins undergo regulation and perform their physiological function. A good example would be a DNA polymerase (see image) and ion channels. Subunits are held together by the same types of interactions that stabilize the tertiary structure of proteins.
There is debate as to whether quaternary structure should be defined to include peptides linked by covalent (disulfide) bonds. In CMB, we will us quaternary structure to refer only to arrangement of subunits that are not covalently linked, although covalent disulfide bonds may occur within the individual subunits.
Understanding Diseases Through Protein Genomics
Cystic Fibrosis
What Is Cystic Fibrosis?
Cystic fibrosis (CF) is a genetic condition that affects a protein in the body. People who have cystic fibrosis have a faulty protein that affects the body’s cells, its tissues, and the glands that make mucus and sweat.
Normal mucus is slippery and protects the airways, digestive tract, and other organs and tissues. Cystic fibrosis causes mucus to become thick and sticky. As mucus builds up, it can cause blockages, damage, or infections in affected organs.
Cystic fibrosis used to cause death in childhood. Survival has improved because of medical discoveries and advances in newborn screening, medicines, nutrition, and lung transplants. Nearly 40,000 children and adults in the United States and more than 100,000 worldwide are now living with cystic fibrosis. Children born between 2018 and 2022 who have cystic fibrosis are expected to live an average of 56 years. On average, half of babies born in 2021 with cystic fibrosis are expected to reach the age of 65 or older.
Some people who have cystic fibrosis have few or no symptoms, while others experience severe symptoms or life-threatening complications. The most serious and common complications of cystic fibrosis are problems with the lungs, including frequent pulmonary or respiratory exacerbations , typically caused by serious lung infections.
Symptoms and Causes
Cystic fibrosis symptoms include:
- Frequent lung infections (recurrent pneumonia or bronchitis).
- Loose or oily poop (stool).
- Trouble breathing.
- Frequent wheezing.
- Frequent sinus infections.
- A nagging cough.
- Slow growth.
- Failure to thrive (inability to gain weight despite having a good appetite and taking in enough calories).
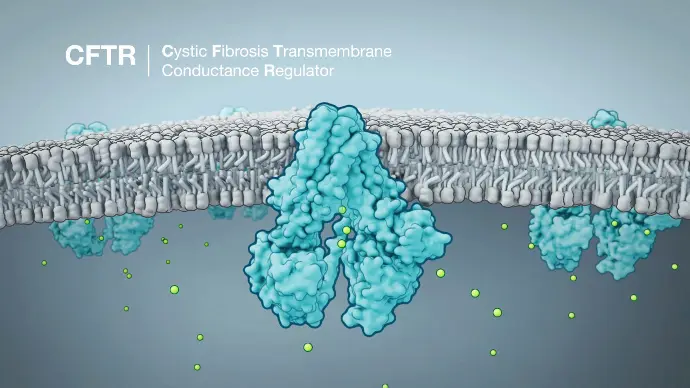
Changes to the CFTR gene — called variants or mutations — cause cystic fibrosis. CFTR makes a protein that works as an ion channel on the surface of a cell. Ion channels are like gates in a cell’s membrane that allow certain molecules to pass through.
CFTR usually makes a gate for chloride ions, a type of mineral with a negative electrical charge. Chloride moves out of the cell, taking water with it, which thins out mucus and makes it more slippery. In people with CF, gene mutations in CFTR prevent this from happening, so the mucus stays sticky and thick.
There are different categories (classes I to VI) of gene mutation in CFTR that depend on the effect they have. Some produce no proteins at all, some produce only small amounts of proteins, and some produce proteins that don’t work properly.
Treatment
An early diagnosis of CF and treatment plan can improve both survival and quality of life. Follow-up and monitoring are very important. When possible, care should be received at a cystic fibrosis specialty clinic. When children reach adulthood, they should transfer to a cystic fibrosis specialty center for adults.
Treatment for lung problems includes:
- Antibiotics to prevent and treat lung and sinus infections. They may be taken by mouth, or given in the veins or by breathing treatments. People with CF may take antibiotics only when needed, or all the time. Doses are often higher than normal.
- Inhaled medicines to help open the airways.
- Other medicines that are given by a breathing treatment to thin mucus and make it easier to cough up are DNAse enzyme therapy and highly concentrated salt solutions (hypertonic saline).
- Flu vaccine and pneumococcal polysaccharide vaccine (PPV) yearly (ask your health care provider).
- Lung transplant is an option in some cases.
- Oxygen therapy may be needed as lung disease gets worse.
Lung problems are also treated with therapies to thin the mucus. This makes it easier to cough the mucus out of the lungs.

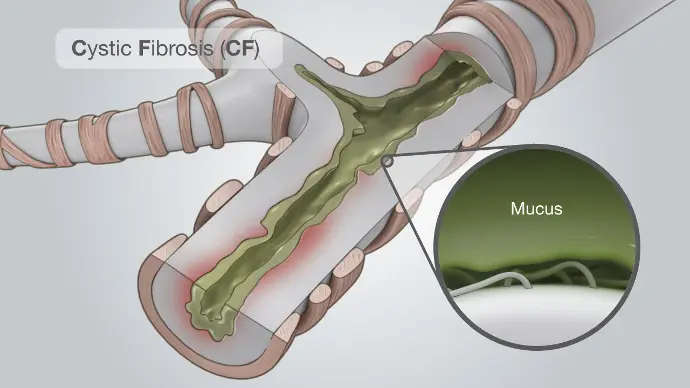
Sickle Cell Disease
Sickle cell disease (SCD) is the most common inherited blood disorder that affects your red blood cells. In SCD, a protein called hemoglobin, located within red blood cells, is abnormal. Hemoglobin is important. It’s the molecule that carries oxygen in your blood and throughout your body.
Normal red blood cells are round and flexible. This allows them to move easily through small blood vessels (capillaries) in your body to deliver oxygen to your organs and tissues. In SCD, there’s an abnormal form of hemoglobin called hemoglobin S. This changes the shape of the red blood cell to a crescent shape and causes red blood cells to become rigid, lack flexibility and stick together. This can block blood flow, preventing oxygen from getting to the vital organs and tissues throughout your body. It can lead to serious complications including pain, infections, and organ damage and failure. Additionally, sickle-shaped cells don’t last as long as normal-shaped red blood cells, causing a constant shortage of red blood cells and leading to anemia. Sickle cell disease is a lifelong condition. But there are treatment options that can reduce your symptoms and prolong your life. Results from a mutation in the gene that codes for hemoglobin, leading to abnormal red blood cells that can cause pain and organ damage.
Causes
SCD is an inherited disease caused by a defect in a gene:
- You are born with SCD only if 2 genes are inherited—1 from each parent.
- If you have just 1 gene you are healthy, but you are a carrier of the disease. If 2 carriers have a child, there is a greater chance their child will have SCD.
- Parents who are each a carrier of a sickle cell gene have a 1 in 4 chance of having a child with SCD.
What are the symptoms of sickle cell disease?
Each person’s symptoms may vary. They may be mild or severe. Symptoms may include:
- Anemia. This symptom is the most common one. Having fewer red blood cells causes anemia. Severe anemia can make you feel dizzy, short of breath, and tired.
- Yellowing of the skin, eyes, and mouth (jaundice). This symptom is common. Sickle cells don’t live as long as normal red blood cells. They die faster than the liver can filter them out. The yellow color is caused by a substance (bilirubin) that is released when the red blood cells die.
- Pain crisis, or sickle crisis. When sickle cells move through small blood vessels, they can get stuck. They can block blood flow and cause pain. The sudden pain can occur anywhere. But it most often happens in the chest, arms, and legs. Blocked blood flow may also cause tissue death.
- Acute chest syndrome. This condition occurs when sickle cells stick together and block oxygen flow in the tiny vessels in the lungs. It can be life-threatening. It often happens suddenly when the body is under stress from infection, fever, or dehydration. Symptoms may seem like pneumonia. They can include fever, pain, and a violent cough.
- Splenic sequestration (pooling). The spleen becomes enlarged and painful when sickle cells get stuck there. Fewer red blood cells are able to move, causing a sudden drop in hemoglobin. It can be deadly if not treated at once.
- Stroke. A stroke is another sudden and severe problem that occurs with this disease. The sickle cells can block the major blood vessels that bring oxygen to the brain. Any interruption in the flow of blood and oxygen to the brain can cause severe brain damage. If you have a stroke from SCD, you are more likely to have a second and third stroke.
- Priapism. The sickle cells block the blood vessels in the penis, causing great pain. If not treated right away, it can cause erectile dysfunction.
The symptoms of SCD may look like other blood disorders or health problems. Always see your healthcare provider for a diagnosis.
Treatments
Early diagnosis and preventing further problems is critical in treating SCD. Treatment goals include preventing organ damage (including strokes), preventing infection, and treating symptoms. Treatment may include:
- Pain medicines. These are used for sickle cell crises.
- Drinking plenty of water daily (8 to 10 glasses). Doing so can prevent and treat pain crises. In some cases, IV (intravenous) fluids may be needed.
- Blood transfusions. These may help treat anemia and prevent stroke. They are also used to dilute the sickled hemoglobin with normal hemoglobin. It is done to treat chronic pain, acute chest syndrome, splenic sequestration, and other emergencies.
- Red blood cell exchange.This process removes some abnormal red blood cells and replaces them with healthy red blood cells from a donor. This can help improve symptoms.
- Vaccines and antibiotics. These are used to prevent infections.
- Folic acid. This B vitamin helps prevent severe anemia.
- Hydroxyurea. This medicine helps reduce the frequency of pain crises and acute chest syndrome. It may also help decrease the need for blood transfusions.
- Voxelotor. This medicine is approved for people age 4 and older to reduce pain crises and improve anemia caused by red blood cell destruction.
- Crizanlizumab. This medicine is approved for people age 16 and older to reduce pain crises.
- L-glutamine. This medicine is approved for people age 5 and older to reduce pain crises.
- Regular eye exams. These are done to screen for an eye condition called retinopathy.
- Bone marrow transplant. A transplant can cure some people with SCD. The decision to have a transplant is based on the severity of the disease and finding a suitable donor. These decisions need to be discussed with your provider. Transplants are done only at specialized medical centers.
Phenylketonuria (PKU)
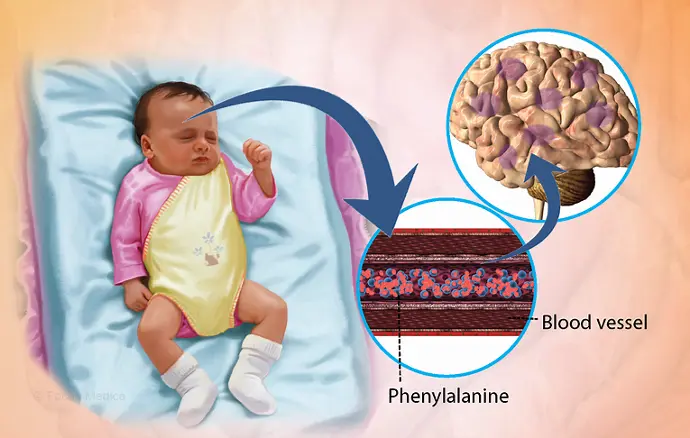
Phenylketonuria , also called PKU, is a rare inherited disorder that causes an amino acid called phenylalanine to build up in the body. PKU is caused by a change in the phenylalanine hydroxylase (PAH) gene. This gene helps create the enzyme needed to break down phenylalanine.
Without the enzyme necessary to break down phenylalanine, a dangerous buildup can develop when a person with PKU eats foods that contain protein or eats aspartame, an artificial sweetener. This can eventually lead to serious health problems.
For the rest of their lives, people with PKU — babies, children and adults — need to follow a diet that limits phenylalanine, which is found mostly in foods that contain protein. Newer medications may allow some people with PKU to eat a diet that has a higher or an unrestricted amount of phenylalanine.
Babies in the United States and many other countries are screened for PKU soon after birth. Although there is no cure for PKU, recognizing PKU and starting treatment right away can help prevent limitations in areas of thinking, understanding and communicating (intellectual disability) and major health problems.
Symptoms
Signs and symptoms of untreated PKU can be mild or severe and may include:
- A musty odor in the breath, skin or urine, caused by too much phenylalanine in the body
- Nervous system (neurological) problems that may include seizures
- Skin rashes, such as eczema
- Lighter skin, hair and eye color than family members, because phenylalanine can't transform into melanin — the pigment responsible for hair and skin tone
- Unusually small head size (microcephaly)
- Hyperactivity
- Intellectual disability
- Delayed development
- Behavioral, emotional and social problems
- Mental health disorders
Causes
A gene change (genetic mutation) causes PKU, which can be mild, moderate or severe. In a person with PKU, a change in the phenylalanine hydroxylase (PAH) gene causes a lack of or reduced amount of the enzyme that's needed to process phenylalanine, an amino acid.
A dangerous buildup of phenylalanine can develop when a person with PKU eats protein-rich foods, such as milk, cheese, nuts or meat, or grains such as bread and pasta, or aspartame, an artificial sweetener.
Treatment
The main treatments for PKU include:
- A lifetime diet with very limited intake of foods with phenylalanine
- Taking a PKU formula — a special nutritional supplement — for life to make sure that you get enough essential protein (without phenylalanine) and nutrients that are essential for growth and general health
- Medications, for certain people with PKU
A safe amount of phenylalanine differs for each person with PKU and can vary over time. In general, the idea is to consume only the amount of phenylalanine that's necessary for healthy growth and body processes, but no more. Your health care provider can determine a safe amount through:
- Regular review of food records and growth charts
- Frequent blood tests that monitor blood phenylalanine levels, especially during childhood growth spurts and pregnancy
- Other tests that assess growth, development and health
Your health care provider may refer you to a registered dietitian who can help you learn about the PKU diet, make adjustments to your diet when needed, and offer suggestions on ways to manage PKU diet challenges
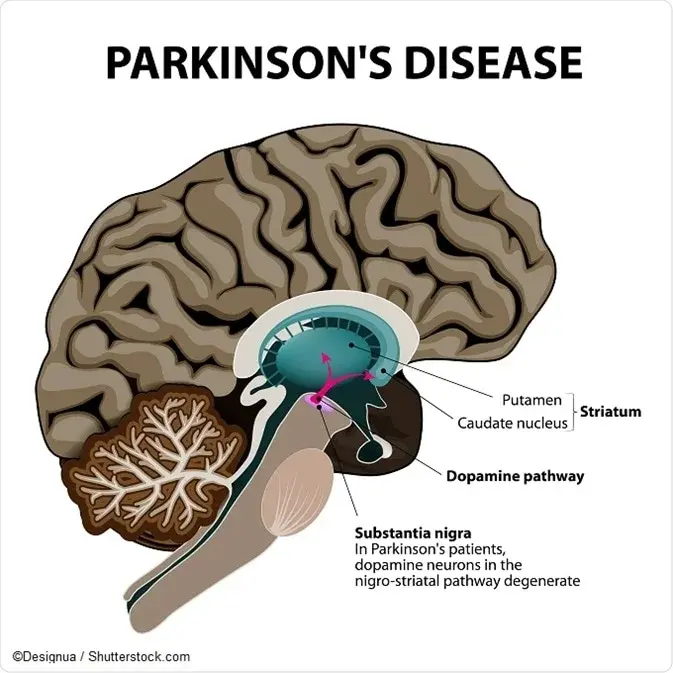
Parkinson's Disease
What is Parkinson's Disease?
Parkinson's disease is a movement disorder of the nervous system that worsens over time. The nervous system is a network of nerve cells that controls many parts of the body, including movement.
Symptoms start slowly. The first symptom may be a barely noticeable tremor in just one hand or sometimes a foot or the jaw. Tremor is common in Parkinson's disease. But the disorder also may cause stiffness, slowing of movement and trouble with balance that raises the risk of falls.
In the early stages of Parkinson's disease, your face may show little or no expression. Your arms may not swing when you walk. Your speech may become soft or slurred. Symptoms get worse over time.
Although Parkinson's disease can't be cured, medicines may help symptoms get better. Sometimes a healthcare professional may suggest surgery to help control parts of the brain. This surgery may help lessen symptoms.
Causes
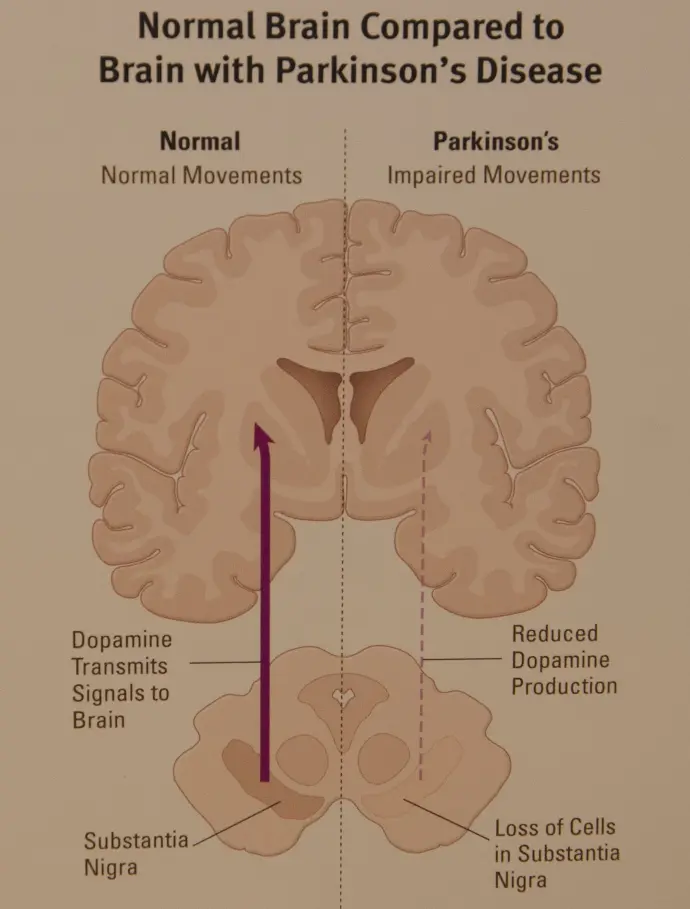
In Parkinson's disease, nerve cells in the brain called neurons slowly break down or die. Many Parkinson's disease symptoms are caused by a loss of neurons that produce a chemical messenger in the brain. This messenger is called dopamine.
Decreased dopamine leads to irregular brain activity. This causes movement problems and other symptoms of Parkinson's disease. People with Parkinson's disease also lose a chemical messenger called norepinephrine that controls many body functions, such as blood pressure.
The cause of Parkinson's disease is unknown, but several factors seem to play a role, including:
- Genes. Specific genetic changes are linked to Parkinson's disease. But these are rare unless many family members have had Parkinson's disease.
- Among familial cases of Parkinson's disease, the inheritance pattern differs depending on the gene that is altered. If the LRRK2 or SNCA gene is involved, the disorder is inherited in an autosomal dominant pattern
, which means one copy of an altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition.
- If the PARK7, PINK1, or PRKN gene is involved, Parkinson's disease is inherited in an autosomal recessive pattern
. This type of inheritance means that both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
- When genetic alterations modify the risk of developing Parkinson's disease, the inheritance pattern is usually unknown.
Symptoms
Parkinson's disease symptoms can be different for everyone. Early symptoms may be mild, and you may not even notice them. Symptoms often begin on one side of the body, then affect both sides. Symptoms are usually worse on one side than the other. Some Parkinson's disease symptoms are similar to those of other disorders.
Parkinson's symptoms may include:
- Tremor. This rhythmic shaking usually begins in the hands or fingers. Sometimes tremor begins in the foot or jaw. You may rub your thumb and forefinger back and forth. This is known as a pill-rolling tremor. Your hand may tremble when it's at rest or when you're under stress. You may notice that you shake less when you're doing some sort of task or moving around.
- Slowed movement, also called bradykinesia. Parkinson's disease may slow your movement, making simple tasks more difficult. It may be hard to get out of a chair, shower or get dressed. You may have less expression in your face. It may be hard to blink.
- Rigid muscles. You may have stiff muscles in any part of your body. Your muscles may feel tense and painful, and your arm movements may be short and jerky.
- Poor posture and balance. Your posture may become stooped. You may fall or have balance problems.
- Loss of automatic movements. You may be less able to make certain movements that you typically do without thinking, including blinking, smiling or swinging your arms when you walk.
- Speech changes. You may speak softly or quickly, slur, or hesitate before talking. Your speech may be flat or monotone, without typical speech patterns.
- Writing changes. You may have trouble writing, and your writing may appear cramped and small.
Treatment
Parkinson's disease can't be cured, but medicines can help control the symptoms. Medicines often work very well. When medicine is no longer helping, some people may have surgery. Your healthcare team also may recommend aerobic exercise, physical therapy that focuses on balancing and stretching, and speech therapy.
Medicines
Medicines may help improve problems with walking, movement and tremor. The medicines work by increasing or substituting for dopamine in the brain.
People with Parkinson's disease have low levels of brain dopamine. But dopamine can't be given directly because it can't enter the brain.
Your symptoms may improve significantly after you start treatment. The benefits may lessen over time, but usually medicines still control symptoms well.
Medicines you may be prescribed include:
- Carbidopa-levodopa (Rytary, Sinemet, others). Levodopa is the most effective Parkinson's disease medicine. It is a natural chemical that passes into the brain and becomes dopamine. Levodopa is combined with carbidopa to help levodopa reach the brain and to prevent or lessens side effects such as nausea.
Side effects may include nausea and lightheadedness when you stand, called orthostatic hypotension. Higher doses of levodopa may cause involuntary movements, known as dyskinesia. If this happens, your dose may need to be lessened or adjusted.
The benefit from levodopa may lessen over time. It also may wax and wane. This is called wearing off.
Carbidopa-levodopa usually is best taken on an empty stomach if you have advanced Parkinson's disease. Follow your healthcare team's advice on the best time to take it. - Inhaled levodopa (Inbrija). This medicine helps manage symptoms when oral medicines suddenly stop working during the day.
- Carbidopa-levodopa infusion (Duopa). This medicine is given through a feeding tube in a gel form and goes directly into the small intestine.
The tube is placed during a minor surgery. There are some risks of having the tube. The tube may fall out or cause an infection at the infusion site.
Carbidopa-levodopa infusion is for patients with advanced Parkinson's who still respond to the medicine but need a more constant level of levodopa. It helps control motor difficulties and other symptoms such as anxiety and depression. - Dopamine agonists. Unlike levodopa, dopamine agonists don't change into dopamine. Instead, they mimic dopamine effects in the brain.